Groups at the Weizmann Institute of Science and Cambridge University have jointly managed the feat of turning back the clock on human cells to create primordial germ cells – the embryonic cells that give rise to sperm and ova – in the lab. This is the first time that human cells have been programmed into this early developmental stage. The results of their study, which were published today in Cell, could help provide answers as to the causes of fertility problems, yield insight into the earliest stages of embryonic development and potentially, in the future, enable the development of new kinds of reproductive technology.
“Researchers have been attempting to create human primordial germ cells (PGCs) in the petri dish for years,” says
Dr. Jacob Hanna of the Institute’s Molecular Genetics Department, who led the study together with research student Leehee Weinberger. PGCs arise within the early weeks of embryonic growth, as the embryonic stem cells in the fertilized egg begin to differentiate into the very basic cell types. Once these primordial cells become “specified,” they continue developing toward precursor sperm cells or ova “pretty much on autopilot,” says Hanna. The idea of creating these cells in the lab took off with the 2006 invention of induced pluripotent stem (iPS) cells – adult cells that are “reprogrammed” to look and act like embryonic stem cells, which can then differentiate into any cell type. Thus several years ago, when researchers in Japan created mouse iPS cells and then got them to differentiate into PGCs, scientists immediately set about trying to replicate the achievement in human cells. But until now, none had been successful.
Previous research in Hanna’s lab pointed to new methods that could take human cells to the PGC state. That research had focused on the question of how human iPS cells and mouse embryonic cells differ: The mouse embryonic cells are easily kept in their stem cell state in the lab, while human iPS cells that have been reprogrammed – a technique that involves the insertion of four genes – have a strong drive to differentiate, and they often retain traces of “priming.” Hanna and his group then created a method for tuning down the genetic pathway for differentiation, thus creating a new type of iPS cell that they dubbed “naïve cells.” These naïve cells appeared to rejuvenate iPS cells one step further, closer to the original embryonic state from which they can truly differentiate into any cell type. Since these naïve cells are more similar to their mouse counterparts, Hanna and his group thought they could be coaxed to differentiate into primordial germ cells.
Working with naïve human embryonic stem and iPS cells, and applying the techniques that had been successful in the mouse cell experiments, the research team managed to produce cells that, in both cases, appeared to be identical to human PGCs. Together with the lab group of Prof. Azim Surani of Cambridge University, the scientists further tested and refined the method jointly in both labs. By adding a glowing red fluorescent marker to the genes for PGCs, they were able to gauge how many of the cells had been programmed. Their results showed that quite a high rate – up to 40% – had become PGCs; this quantity enables easy analysis.
Hanna points out that PGCs are only the first step in creating human sperm and ova. A number of hurdles remain before labs will be able to complete the chain of events that move an adult cell through the cycle of embryonic stem cell and around to sperm or ova. For one, at some point in the process, these cells must learn to perform the neat trick of dividing their DNA in half before they can become viable reproductive cells. Still, he is confident that those hurdles will one day be overcome, raising the possibility, for example, of enabling women who have undergone chemotherapy or premature menopause to conceive.
In the meantime, the study has already yielded some interesting results that may have significant implications for further research on PGCs and possibly other early embryonic cells. The team managed to trace part of the genetic chain of events that directs a stem cell to differentiate into a primordial germ cell, and they discovered a master gene, Sox17, that regulates the process in humans, but not in mice. Because this gene network is quite different from the one that had been identified in mice, the researchers suspect that more than a few surprises may await scientists who study the process in humans.
Hanna: “Having the ability to create human PGCs in the petri dish will enable us to investigate the process of differentiation on the molecular level. For example, we found that only ‘fresh’ naïve cells can become PGCs; but after a week in conventional growth conditions they lose this capability once again. We want to know why this is. What is it about human stem cell states that makes them more or less competent? And what exactly drives the process of differentiation once a cell has been reprogrammed to its more naïve state? It is the answers to these basic questions that will, ultimately, advance iPS cell technology to the point of medical use.”
This collaborative project was made possible by a grant from BIRAX Britain Israel Research and Academic Exchange Partnership – Regenerative Medicine Initiative.
Dr. Jacob Hanna’s research is supported by Pascal and Ilana Mantoux, France/Israel; the New York Stem Cell Foundation, the Flight Attendant Medical Research Institute (FAMRI), the Israel Cancer Research Fund (ICRF), the Helen and Martin Kimmel Award for Innovative Investigation, the Benoziyo Endowment Fund for the Advancement of Science; the Leona M. and Harry B. Helmsley Charitable Trust; the Sir Charles Clore Research Prize; Erica A. Drake and Robert Drake; the Abisch Frenkel Foundation for the Promotion of Life Sciences; the European Research Council; the Israel Science Foundation, and the Fritz Thyssen Stiftung. Dr. Hanna is a New York Stem Cell Foundation-Robertson Investigator.

















 Hanna")


 state (colored cells). Each color reveals the expression of a different gene. (l-r) NANOS3, NANOG, OCT4 and, on the right, all three combined in a single image")
 state (colored cells). Each color reveals the expression of a different gene. (l-r) NANOS3, NANOG, OCT4 and, on the right, all three combined in a single image")

 in tRNA signatures among cancerous cells and healthy dividing cells, as well as a degree of similarity among the non-dividing cells (red colors, lower left rectangle), whereas no such similarity (blue) was found when dividing cells were compared with non-dividing ones")
 in tRNA signatures among cancerous cells and healthy dividing cells, as well as a degree of similarity among the non-dividing cells (red colors, lower left rectangle), whereas no such similarity (blue) was found when dividing cells were compared with non-dividing ones")

, is found in the same cellular area as Presenilin, around the nucleus")


 divulge the wealth of interconnections between two major cell death pathways")

 divulge the wealth of interconnections between two major cell death pathways")




 Idan Frumkin, Zohar Bloom-Ackerman, Prof. Tzachi Pilpel, Dr. Orna Dahan and Avihu Yona")




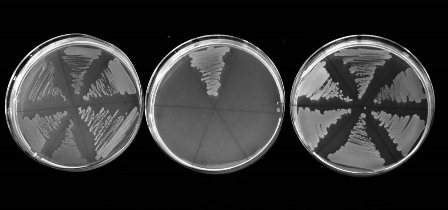
%20(2).jpg "Growth of bacteria when only antitoxin is induced (left), only toxin is induced (middle), and both are induced together (right)")



Use It or Lose It
This signal presumably alters the internal architecture of the chromosomes within the nucleus, changing gene expression – that is, the activity of genes – in the affected portion of the DNA. As a result, certain genes become activated, prompting the release of proteins that make up the filaments responsible for the contraction of the muscle fiber. In addition to contraction, the proteins fortify existing filaments and help produce new ones, building up muscle mass.
Evidence for this explanation comes from a series of studies that Volk and her team – including research students Hadas Elhanany-Tamir and Miri Shnayder, and postdoctoral fellow Dr. Shuoshuo Wang – conducted in fruit fly larvae. As reported in the Journal of Cell Biology, the scientists have identified a fruit fly protein, called MSP-300, whose shape and mechanical properties make it perfectly suited to serving as a mechanosensor. MSP-300 forms a ring around the nucleus, with numerous radial extensions connected to the cytoskeleton. It has elastic properties, as would be required from a mechanosensor; but on the other hand, it operates in close cooperation with two other proteins that help create rigid scaffolding around the nucleus, protecting it from muscle contractions. By introducing mutations into MSP-300, the scientists showed that it is indeed essential for maintaining muscle mass. The effect of the mutations on the fly’s muscles was devastating: The muscle fibers thinned out and their nuclei were deformed and clumped together abnormally. As a result, the muscles didn’t work properly, so the larvae couldn’t crawl and adult flies couldn’t fly.
Because human muscle cells contain proteins equivalent to MSP-300, suggesting they may also function as mechanosensors, these findings can shed new light on the connection between physical activity and a healthy build-up of muscle in humans. A better understanding of this connection may in the future lead to improved ways to prevent muscle loss resulting from aging, forced inactivity as in paralysis, or such disorders as muscular dystrophy.
Prof. Talila Volk's research is supported by Erica A. Drake and Robert Drake. Prof. Volk is the incumbent of the Sir Ernst B. Chain Professorial Chair.